能量分析
4. 轨迹分析
使用以下命令在human(suprerim/docking)文件夹下新建md_analysis文件夹,并进入该文件夹内:
$cd $cd human $mkdir md_analysis $cd md_analysis
使用Vim编辑器在md_analysis目录下新建以下文件进行轨迹的分析:
注意!在对superim与docking进行分析时,以下标红的部分应进行相应的更改(如下面的文件名应分别改为cpptraj_superim_ligandPositionalRMSD.in 与cpptraj_docking_ligandPositionalRMSD.in)
1.轨迹动画
MD模拟的轨迹实际上只是一系列顺序相关的快照,因此可以将其视为动画。 我们将使用VMD以实际的影片格式渲染轨迹。
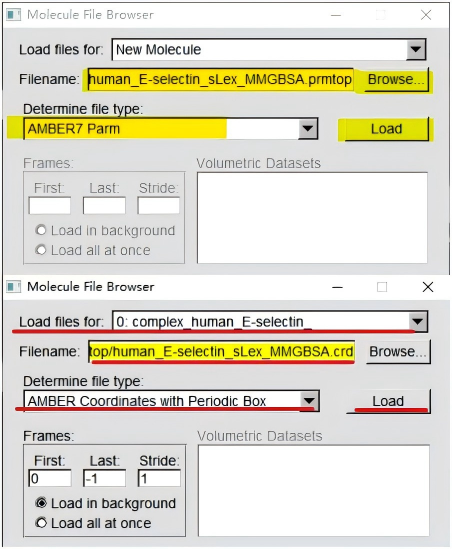
首先,需要通过Filezilla复制无水文件complex_human_E-selectin_sLex_MMGBSA.prmtop和轨迹无水文件human_E-selectin_sLex_MMGBSA.crd文件(这两个文件在human/docking/superim文件夹下的md_calculate文件夹下)到本地计算机。
在本地PC上,打开VMD(基本操作和安装包见VMD部分).
 |
 |
 |
|
 |
|
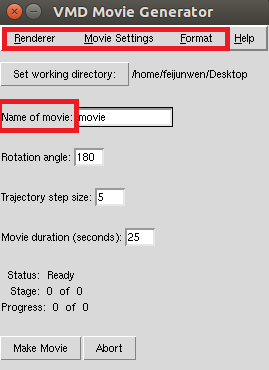 |
完成以上设置后,开始制作动画。打开Extensions->Visualization->Movie Maker功能面板,在该面板Movie Generator中,修改以下参数:
修改完后点击Make Movie 开始制作轨迹动画。该动画可以用在小组PPT展示上 |
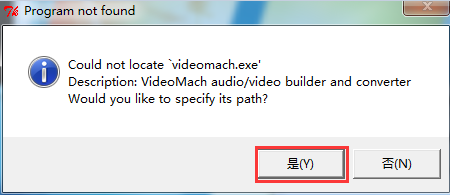 |
如左侧所示的窗口会出现,点击yes即可。 |
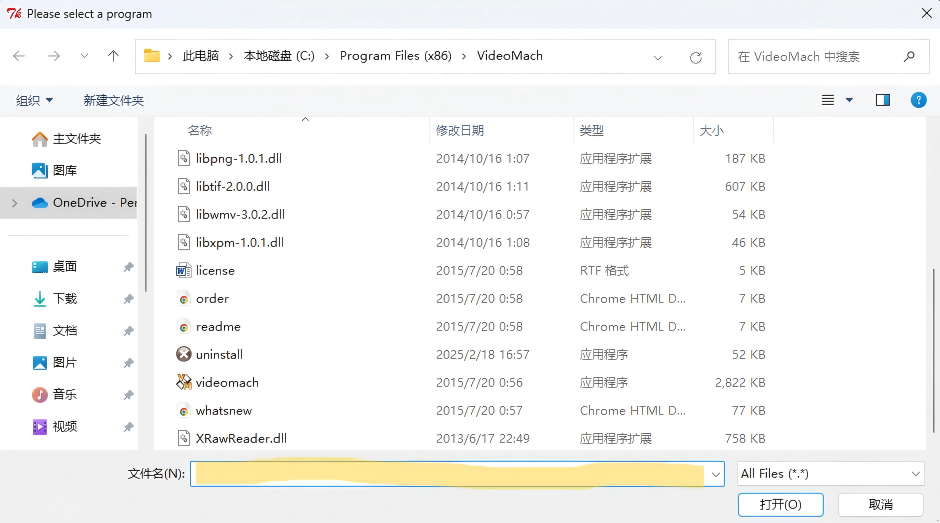 |
在标记处输入如下的VideoMach路径并点击 open(O). C:\program Files(x86)\ViedoMach\vediomach.exe |
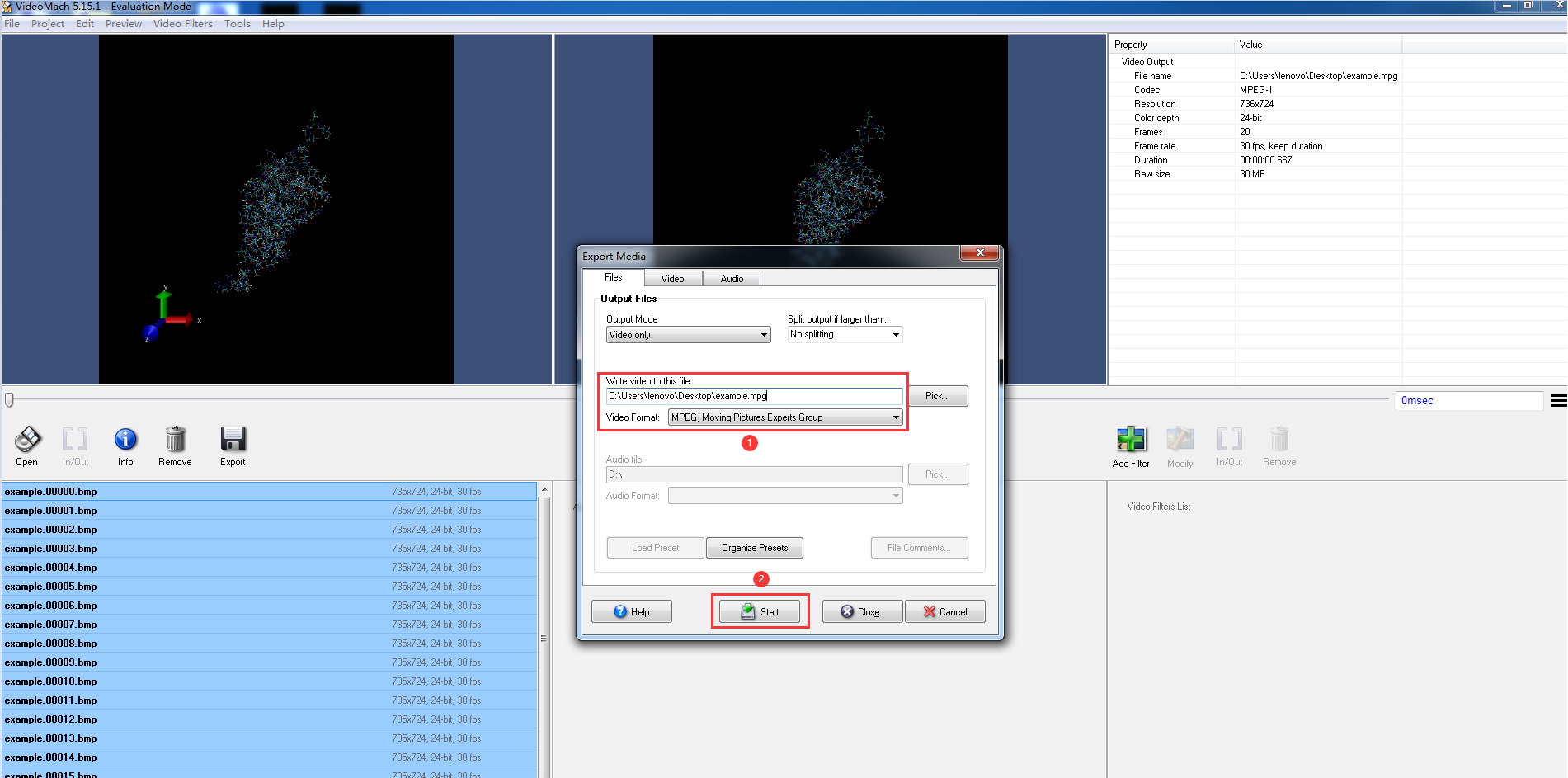 |
然后你会看到VideoMach界面和一个弹出窗口。将红色框中的路径更改为您想要的文件名 接着,点击 start. |
 |
最后您将看到一个弹出窗口,如左侧所示,单击 close. 现在就能看到制作的video出现在桌面上了。 |
2.提取能量
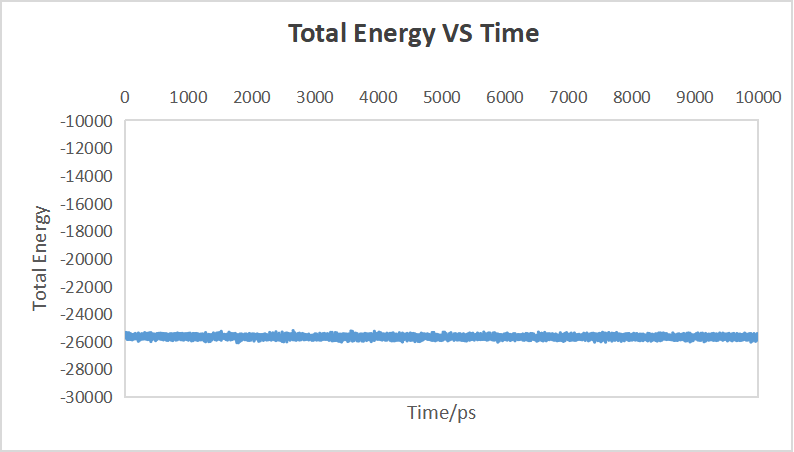
这里我们将提取MD输出文件中的能量和相关数据并作图以观察能量在模拟过程中的变化
在服务器终端上,切换到md_analysis目录并输入以下命令:
$ grep Etot ../10.produ.o > Etot_time.txt
同样的将Etot_time.txt文件传输到本地,并利用Excel作图,这里我们选取变量为Etot 和 Frame,做散点图,选择平滑曲线.
3.提取温度
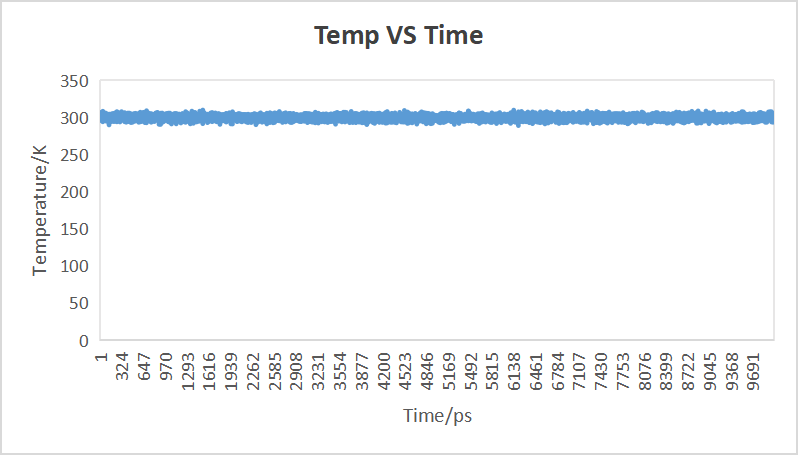
这里我们将提取MD输出文件中的温度和相关数据并作图以观察温度在模拟过程中的变化
切换到md_analysis目录并输入以下命令:
$ grep TIME ../10.produ.o > temp_time.txt
同样的将temp_time.txt文件传输到本地,并利用Excel作图,这里我们选取变量为Temperature 和 Frame.
4.氢键分析
新建以下文件进行氢键的分析
cpptraj_human_Hbond.in
parm ../complex_human_E-selectin_sLex.prmtop trajin ../produ_complex_human_E-selectin_sLex_WAT.nc hbond hbonds out human_E-selectin_sLex_HBONDS.dat angle 120 dist 3.5 avgout human_E-selectin_sLex_HBONDS-Avg.dat nointramol go
在终端运行以下命令:
$cpptraj -i cpptraj_human_Hbond.in
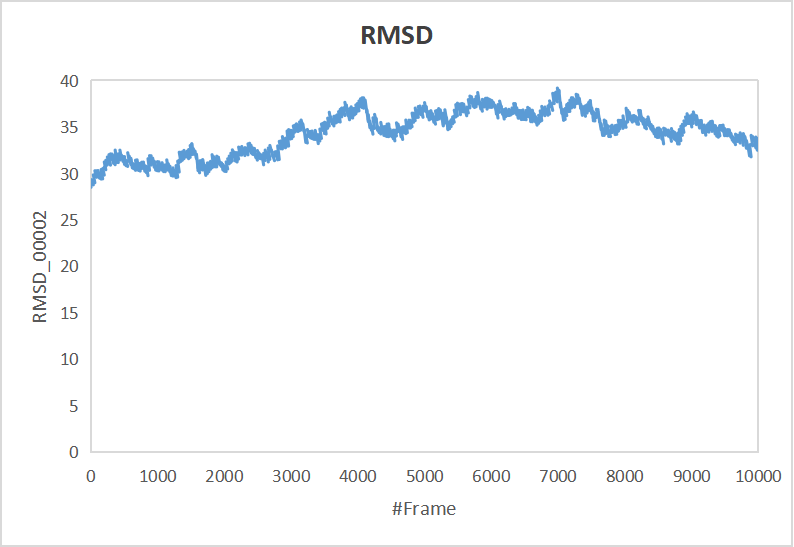
5.计算RMSD
在md_anlysis目录下新建以下文件:
cpptraj_human_ligandPositionalRMSD.in
parm ../complex_human_E-selectin_sLex.prmtop trajin ../produ_complex_human_E-selectin_sLex_WAT.nc reference ../complex_human_E-selectin_sLex.rst7 rms reference out human_E-selectin_sLex_ligand_positionalRMSD.txt :122-126 nofit go
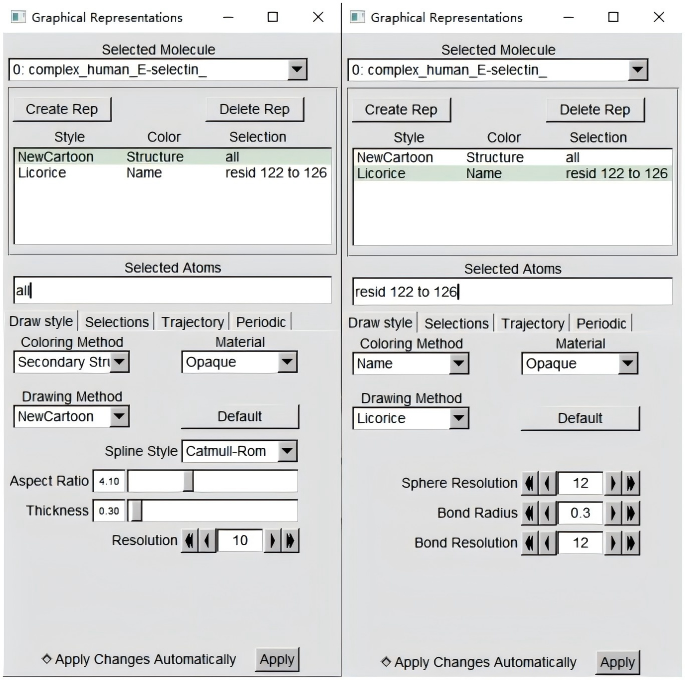
更改标红参数为配体残基范围
在终端运行命令:
$cpptraj -i cpptraj_human_ligandPositionalRMSD.in
将human_E-selectin_sLex_ligand_positionalRMSD.txt移到本地,用excel打开,使用带平滑线的散点图做图
6.计算平均结构
首先,我们将使用cpptraj根据在仿真过程中观察到的所有不同结构来计算平均结构。 请注意,平均结构不是真实的!
切换到md_analysis目录下。在服务器终端上,使用vi编辑get_avg_struct.in文件.
$ vi get_avg_struct.in
将以下文本粘贴到get_avg_struct.in文件中,然后将红色的残基范围编辑为配体的正确残基范围。 将文件保存到md_analysis目录。(请注意修改你的残基范围,包含复合物与离子,但不包含水,这里是1-126)
# Get average structure
parm ../complex_human_E-selectin_sLex.prmtop
trajin ../produ_complex_human_E-selectin_sLex_WAT.nc
average human_traj_avg_struct.pdb :1-126 pdb
run
exit
通过以下命令来运行.(注意这里我们使用的是去掉水的top文件)
$ cpptraj -i get_avg_struct.in
7.挑选代表结构
接下来,我们将根据模拟过程中的平均结构来计算RMSD。 由此可见,MD的哪个结构是平均结构的最佳近似。 这样,我们得到一个真实的“平均”结构。
在服务器终端上,使用vi编辑rmsd_from_avg.in.(请注意修改你的残基范围,仅包含配体,这里是122-126)
$ vi rmsd_from_avg.in
将以下文本粘贴到ptraj_rmsd_from_avg.in,注意修改对应的残基范围
在服务器终端上,使用vi编辑rmsd_from_avg.in.(请注意修改你的残基范围,仅包含配体,这里是122-126)
# Calculate RMSD from average structure
parm ../complex_human_E-selectin_sLex.prmtop
trajin ../produ_complex_human_E-selectin_sLex_WAT.nc
reference ./human_traj_avg_struct.pdb
rms reference out rmsd_from_avg.txt :122-126
run
exit
通过以下命令来运行
$ cpptraj -i rmsd_from_avg.in
将rmsd_from_avg.txt文件传输到本地并作图。然后找到最低点 (这里为 2721),记住该数字,这就是你的最终代表结构的帧数,下一步我们将导出该结构
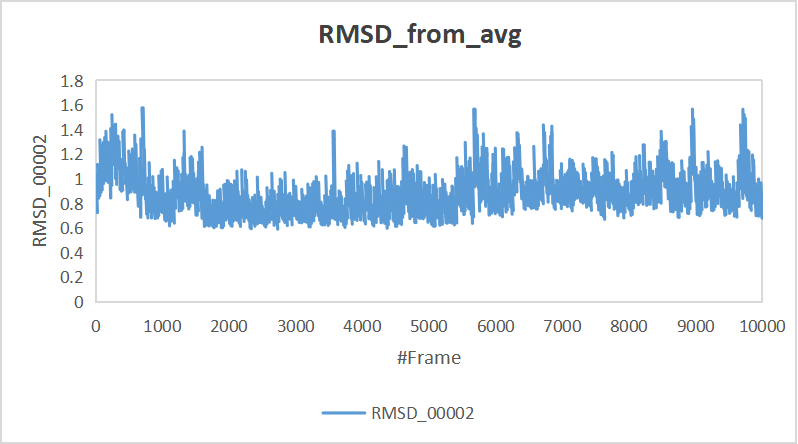
最后,我们将基于以上获得的平均结构和RMSD值来导出真实的代表构象
在服务器终端上,使用vi编辑get_refine_struct.in。将以下文本粘贴到get_refine_struct.in,并修改对应的数字。
#Get snapshot most like the average structure
parm ../complex_human_E-selectin_sLex.prmtop
trajin ../produ_complex_human_E-selectin_sLex_WAT.nc 2721 2721 1
trajout human_struct_like_avg.pdb pdb
run
exit
通过以下命令来运行。
$ cpptraj -i get_refine_struct.in
得到的struct_like_avg.pdb即目标结构。